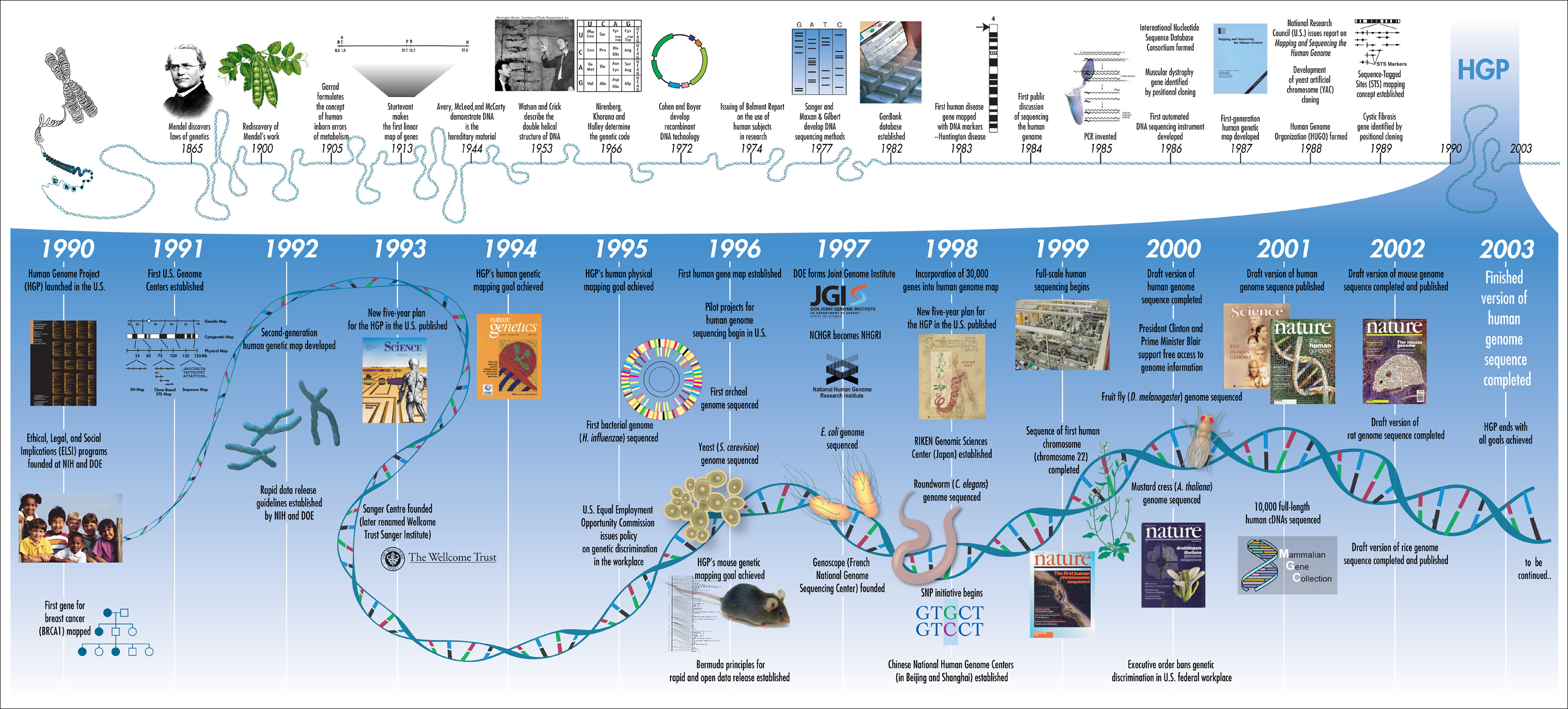
L’immagine sopra rappresenta le tappe fondamentali per la scoperta del genoma umano. Una versione più interattiva della mappa è disponibile nel sito del progetto genoma umano, nella sezione dedicata alla divulgazione.
Perchè abbiamo deciso di sequenziare il genoma umano
Le cellula del nostro corpo, sebbene facciano cose diverse una dall’altra (ad esempio una cellula nervosa che porta il messaggio di muoversi ad una cellula del muscolo che a sua volta si contrae sostenuta dalle cellula dell’osso e dei tendini a cui è vicina) contengono tutte una stessa identica sequenza di DNA, il nostro genoma. Il genoma costituisce l’informazione necessaria e sufficiente per farci essere così come siamo ( o almeno la stragrande maggioranza del come siamo, poi si accumulano anche i segni del tempo e dell’ambiente circostante..).
L’informazione nel nostro genoma è data dalla sequenza con la quale si succedono i 4 nucleotidi Adenina, Timina, Citosina e Guanina.
Le molecole all’interno delle nostre cellule sono in grado di capire il significato delle varie sequenze e tradurlo in funzioni. Così le cellule del nostro corpo appaiono così come le osserviamo e portano avanti compiti estremamente complicati come quelli che ci servono per osservare e capire le cellule del nostro stesso corpo.
Quando studiamo il genoma umano vogliamo cercare di capire, esattamente come fanno le nostre molecole, il significato di quelle sequenze per capire cosa dice il nostro programma e come funziona. Se avessimo capito tutto da un DNA trovato sulla scenza di un delitto, potremmo ricostruire l’identikit completo di una persona.. e invece sappiamo ancora così poco che non sapremmo da dove cominciare!
Molti di voi hanno detto che se capissero il genoma umano vorrebbero usarlo per capire meglio le malattie che ci affliggono e cercare di curarle, molti di noi cercano nel DNA non solo le risposte alle tante domande che abbiamo ma anche le nuove domande che non eravamo neanche in grado di immaginare prima di dare un’occhiata al nostro intero DNA. E infatti il sequenziamento genoma umano ha dischiuso tanti di quei nuovi interrogativi che ha dato origine ad un nuovo progetto che si chiama “Encode” un progetto enciclopedico per mettere insieme tutte le informazioni che troviamo e poi costruire una mappa, una teoria coerente di tutto quello che abbiamo visto e continuiamo a vedere!
Come abbiamo fatto e come facciamo oggi a sequenziare il genoma umano
Quando il progetto genoma umano è iniziato conoscevamo un solo metodo di sequenziamento, chiamato metodo Sanger, dal nome del suo scopritore. Una serie di video pubblicati nella sezione didattica del sito internet del progetto genoma umano spiegano come funziona questo metodo in una delle versioni più moderne. Si tratta delle animazioni da n 6 a n 9 in questa pagina web: http://www.genome.gov/25019885. (Sul libro “Dal carbonio agli OGM PLUS” edito da Zanichelli, a pagina 179 viene spiegato il sequenziamento Sanger).
Nel metodo Sanger un enzima che è in grado di “leggere” le sequenze di DNA, la DNA Polimerasi, viene utilizzato per leggere le sequenze al posto nostro. Dobbiamo quindi trovare un mezzo di comunicazione fra l’enzima e noi e un mezzo per amplificare il segnale di questa comunicazione in modo tale da essere capaci di percepire il messaggio.
La strategia che trovò Sanger fu quella di lasciare che la polimerasi sintetizzasse un filamento di DNA complementare a quello di un filamento sconosciuto di cui vogliamo conoscere la sequenza e di introdurre alcuni nucleotidi speciali in grado di interrompere la sintesi del nuovo filamento. Ciascuno di questi nucleotidi era marcato nella versione più moderna da una molecola fluorescente, pertanto ogni volta che la sequenza si interrompe in una certa posizione sappiamo dire qual’è il nucleotide in quella posizione in base al colore della fluorescenza che emette (un colore diverso per ogni nucleotide). Ma come facciamo a sapere in che posizione si è fermata la polimerasi? Se tutte le sequenze partono dalla stessa posizione basta dividere i frammenti che otteniamo per dimensione e metterli in fila in modo tale da ottenere un frammento per ogni posizione (uno lungo 1, uno lungo 2. uno lungo 3 e così via) e poi guardare il colore di ciascun frammento (solo il nucleotide che fa fermare la polimerasi è colorato, tutti quelli precedenti non emettono nessuna fluorescenza). Il modo migliore per capire è guardare i video! Le sequenze ottenute con il metodo Sanger sono lunghe al massimo 1000 paia di basi.
Oggi abbiamo molti metodi diversi per ottenere la sequenza di un frammento di DNA, ma tutti i metodi usano questa regola generale: c’è uno o più enzimi che leggono la sequenza per noi, ci siamo inventati un sistema di comunicazione fra noi e gli enzimi e un sistema per amplificare il messaggio.
Tutti i metodi che conosciamo oggi si possono fare in modo automatico utilizzando macchine in grado di far avvenire tutti i passaggi della reazione di sequenziamento e le macchine sono controllate da computer.
Tutti i metodi che conosciamo danno sequenze più o meno lunghe, ma sempre di lunghezza limitata. Danno un numero più o meno grande di sequenze in un tempo più o meno breve e ad un costo più o meno basso. Ciascun metodo ha pregi e difetti diversi e pertanto è utile per applicazioni diverse oppure si possono applicare diversi metodi alla stessa sequenza ed integrare i risultati. In ogni caso oggi possiamo ottenere l’intera la sequenza del genoma di un uomo in poche ore al costo minimo di 1000 dollari.
Se le sequenze sono in ogni caso di lunghezza limitata, dovremo sempre avere un metodo per mettere i pezzettini di sequenze insieme ed assemblare i cromosomi. Per farvi un’idea di come questo si faccia provate a fare questo giochino: nell’immagine qui sotto c’è una frase scomposta in parole casuali lunghe 5 lettere. Prendiamo 114 di queste parole casuali e cerchiamo di metterle insieme per ricostruire la frase originale. Ci riuscite? vedrete che più aumentiamo il numero e/o la lunghezza delle parole più è facile. Inoltre vedrete che quando una parola è ripetuta più volte nella frase è sempre difficile capire dove metterla, a volte è impossibile. Esattamente questo succede quando mettiamo insieme le sequenze per assemblare i cromosomi.
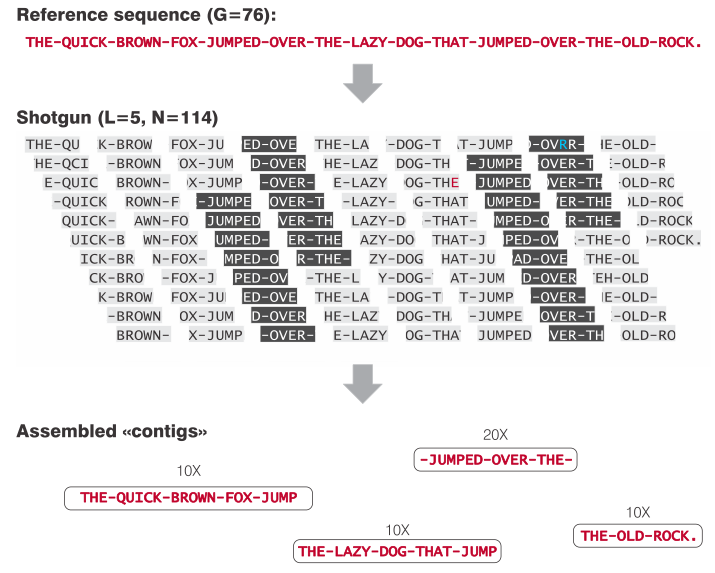
La pubblicazione di questa immagine è una cortesia del Dottor Andrea Telatin
La cosa importante è dare un senso alle sequenze che abbiamo ottenuto
Una volta ottenute le sequenze dei nostri cromosomi la cose che vogliamo scoprire sono:
- ci sono geni che codificano per proteine o RNA?
- qual’è la funzione di queste proteine e di questi RNA?
- ci sono differenze fra individuo e individuo? e se si cosa comportano? quali sono le differenze di un individuo con una certa malattia rispetto ad un individuo sano per quella stessa malattia?
Ovviamente le nostre domande sono infinite, ma queste sicuramente sono le più fondamentali e le più immediate come avete suggerito anche voi durante le nostre lezioni.
Per trovare la risposta a queste domande ci avvaliamo di una idea molto semplice ma molto potente: se il significato di pezzetto di DNA è dato dalla sequenza delle sue basi, e rappresento ciascuna base con una lettera, quel pezzetto di DNA altro non sarà che una parola e un pezzo di DNA più grande altro non sarà che un testo formato da tante parole (per approfondire questo concetto potete svolgere l’esercitazione di introduzione alla bioinformatica, richiede circa 2 ore e può essere fatta da qualsiasi studente che abbia un computer e una connessione a internet). Possiamo analizzare questo testo usando vari criteri e le operazioni che facciamo le possiamo codifcare in un programma e farle svolgere ad un computer al posto nostro.
Per trovare i geni che codificano per proteine proveremo a leggere le sequenze a gruppi di tre lettere partendo da tutte le posizioni possibili e a tradurre queste triplette in aminoacidi. Ogni volta che il numero di aminoacidi che otteniamo uno di seguito all’altro senza stop supera 50, possiamo presumere che quella regione del DNA codifichi verosimilmente per una proteina. (per capire meglio e approfondire, leggete questa spiegazione corredata da figure http://www.genome.gov/25020001)
Se vogliamo scoprire quale sia la funzione delle proteine che abbiamo “predetto” traducendo le triplette trovate sulla sequenza del genoma, il modo migliore per il momento è confrontare le sequenze di queste proteine con quelle di altre proteine già studiate e vedere se si somigliano. Se due proteine hanno una sequenza molto simile probabilmente hanno anche la stessa forma e se la sequenza è molto molto simile probabilmente non solo la forma ma anche la funzione sarà la stessa (per capire meglio e approfondire, leggete questa spiegazione corredata da figure http://www.genome.gov/25020002)
Infine se vogliamo trovare le differenze fra sequenze di individui diversi ancora una volta dovremo confrontare fra di loro le sequenze di questi due individui, vedere se ci sono differenze e una volta trovate queste differenze vedere se si trovano nella porzione di DNA che codifica per una proteina ed eventualmente per una proteina con quale funzione. (per capire meglio e approfondire, leggete questa spiegazione corredata da figure http://www.genome.gov/25020003)
Per capire bene in pratica i tre passaggi che vi ho appena illustrato, potete provare voi stessi l’esercizio sui batteri che provocano la tubercolosi nell’esercitazione di introduzione alla bioinformatica.
A questo link trovate una versione stampabile della lezione. Per favore chiedetevi se vi serva veramente stamparla prima di sprecare la carta!





ciao mi manderesti la lezione in ppt