
Facciamo una PCR “in silico” per isolare la sequenza Alu di nostro interesse
Cliccate su questo link per accedere al programma BLAST. Questo programma vi consente di confrontare fra loro delle sequenze biologiche e di confrontarle con tutte le sequenze presenti nella banca dati internazionale delle sequenze note.
Facciamo una PCR virtuale cercando le sequenze dei nostri primer di interesse sulle sequenze di genoma umano presenti in banca dati.
Queste sono le sequenze dei primer che abbiamo usato nel nostro esperimento:
5′- GGATCTCAGGGTGGGTGGCAATGCT-3′ (forward primer )
5′- GAAAGGCAAGCTACCAGAAGCCCCAA-3′ (reverse primer )
Nella pagina iniziale del programma Blast scegliete la versione utile a confrontare i nucleotidi fra loro (Nucleotide Blast).
Quando sarete nell’interfaccia “Blast n” incollate le sequenze dei primer nella finestra “Enter Query Sequence” facendo attenzione ad incollare le sole sequenze di lettere che rappresentano i nucleotidi (eliminate 5′- e -3′ ) una di seguito all’altra senza lasciare spazi vuoti.
Scegliete di confrontarli con l’intera banca dati delle sequenze nucleotidiche selezionando nel menu a tendina accanto alla parola “Database” l’opzione “Nucleotide collection (nr/nt)”
Scegliete inoltre di cercare sequenze simili ma non identiche (dato che in mezzo fra i due primer ci aspettiamo di trovare altri nucleotidi) selezionando nella sezione “Program Selection” l’opzione “Somewhat similar sequences (blastn)”
A questo punto cliccate sul pulsante azzurro BLAST in basso a sinistra .. e guardiamo insieme il risultato:
Il grafico riassuntivo dei risultati di Blast
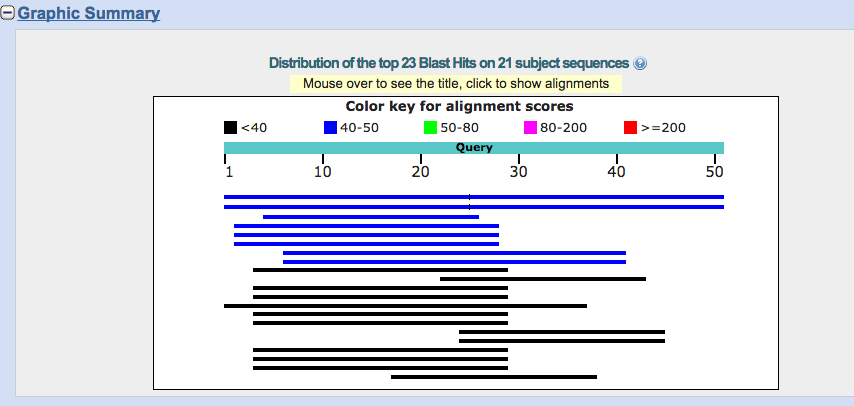
Un codice di colori indica la lunghezza degli allineamenti ottenuti e il grafico a barre mostra l’estensione degli allineamenti fra le sequenze trovate in banca dati e la sequenza “query” (ovvero quella che abbiamo chiesto di cercare). Nelle prime due barre si vede anche che c’è un’interruzione (una lineetta nera interrompe la barra blu), l’allineamento non è continuo.
I parametri statistici degli allineamenti ci consentono di dare un significato ai risultati che abbiamo ottenuto

Query cover indica la percentuale di sequenza query che è stata allineata su una sequenza presente nella banca dati
E value indica la probabilità che quel risultato di allineamento sia imputabile al caso
Ident indica la percentuale di identità fra le due sequenze allineate
Analisi degli allineamenti significativi
Selezioniamo ora e analizziamo uno per volta gli allineamenti significativi cliccando sul nome della sequenza in blu oppure facendo scorrere la pagina.
Leggiamo con attenzione lo schema riassuntivo delle informazioni riguardanti la sequenza presente in banca dati che si allinea alla nostra e i guardiamo i dettagli dell’allineamento

- l’allineamento è compatibile con quanto ci si aspetta da una coppia di primer?
- Sareste in grado di calcolare la lunghezza dell’amplificato?
- Su quale cromosoma umano si allineano i primer?
- La regione in cui si allineano i primer contiene un gene? Se sì, quale?
Ricaviamo la sequenza
Sullo schema riassuntivo dell’allineamento è possibile cliccare su “Genbank” e “Graphics”, iniziamo da “Graphics”
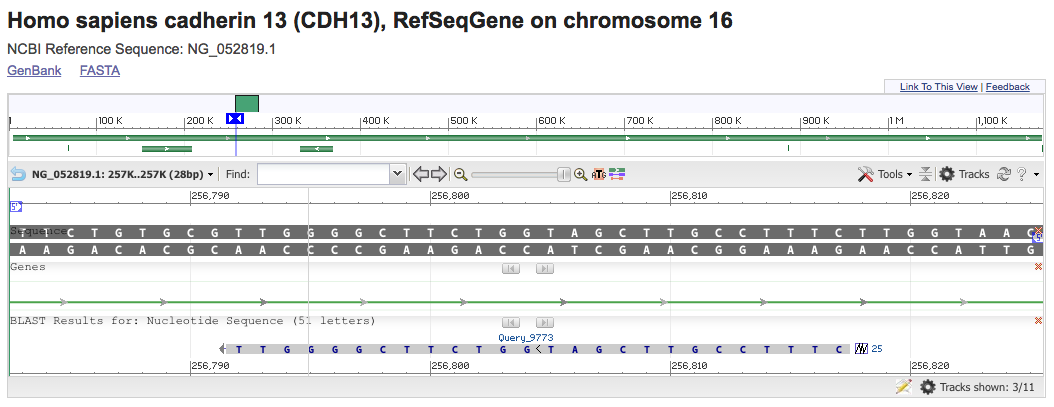
Cliccando su “Graphic” ottenete la possibilità di visualizzare la porzione di sequenza di genoma umano che avete ritrovata con blast su una barra di scorrimento. Oltre alla sequenza sono presenti nel grafico le annotazioni, cioè le informazioni di cui disponiamo su quel tratto di DNA (geni, possibili trascritti, profili di espressione dei trascritti..)
Come prima cosa familiarizzate con lo zoom e andate a vedere il tratto su cui si allineano entrambi i primer:
- uno zoom intorno al 70% dovrebbe essere utile per visualizzare comodamente la posizione e l’orientamento di entrambi i primer e per vedere l’amplificato
da questa posizione potete evidenziare l’area che vi interessa con il mouse
 e un menù a tendina che compare cliccando sulla selezione vi darà la possibilità di salvarla su un file.
e un menù a tendina che compare cliccando sulla selezione vi darà la possibilità di salvarla su un file.Ecco fatta la vostra PCR virtuale con tanto di dettagliata sequenza a disposizione!
Ritrovate il file nel vostro computer (di solito viene scaricato nella cartella “downloads” e cambiategli nome ed estensione: scegliete voi un nome che ritenete descrittivo del file, imponete come estensione .txt
Aprite il file e leggerete la sequenza della vostra PCR! Tenete il file a disposizione perchè lo riuseremo fra poco!
- uno zoom intorno al 5-10% dovrebbe consentirvi di visualizzare le annotazioni di quel tratto di DNA. In particolare c’è un lungo gene, indicato in verde all’interno del quale cade il nostro allineamento
- Sapresti dire di che gene si tratta?
- Quanti esoni e quanti introni ha?
- Il nostro allineamento cade all’interno di un esone o di un introne? Quale?
- Quali altri elementi sono annotati in quella zona?
A questo link trovi un report completo con rappresentazione grafica dell’annotazione del gene che. E’ interessante guardare insieme le informazioni.
La sequenza che abbiamo trovato sul genoma
La PCR virtuale cha avete ottenuto è l’amplificato fra i vostri due primer che si otterrebbe se stessimo utilizzando il genoma di riferimento, cioè un genoma modello. Come sappiamo però, alcuni soggetti hanno l’inserto ALU fra quei due primer, altri no. Come facciamo a sapere se quell’amplificato contiene la sequenza Alu o meno? Si tratterà dell’allele + o dell’allele -?
Caccia alle sequenze Alu PV92 nel genoma umano!
Recuperiamo la nostra PCR virtuale e andiamo a cercarla nella banca dati, vediamo se ci sono sequenze simili a lei:
- apri il file in cui hai salvato la tua pcr virtuale, seleziona tutto e copia
- clicca su questo link per accedere al programma blast e incolla la tua sequenza nella finestra query
- scegli nuovamente nella sezione “Program Selection” l’opzione “Somewhat similar sequences (blastn)”
- clicca sul bottone blast e attendi di vedere i risultati
Anche questa volta osserviamo il grafico riassuntivo degli allineamenti e i parametri statistici degli allineamenti. Ci sono novità interessanti rispetto alla ricerca precedente? (occhio ai trattini verticali neri sulle barrette che rappresentano un’interruzione nell’allineamento)
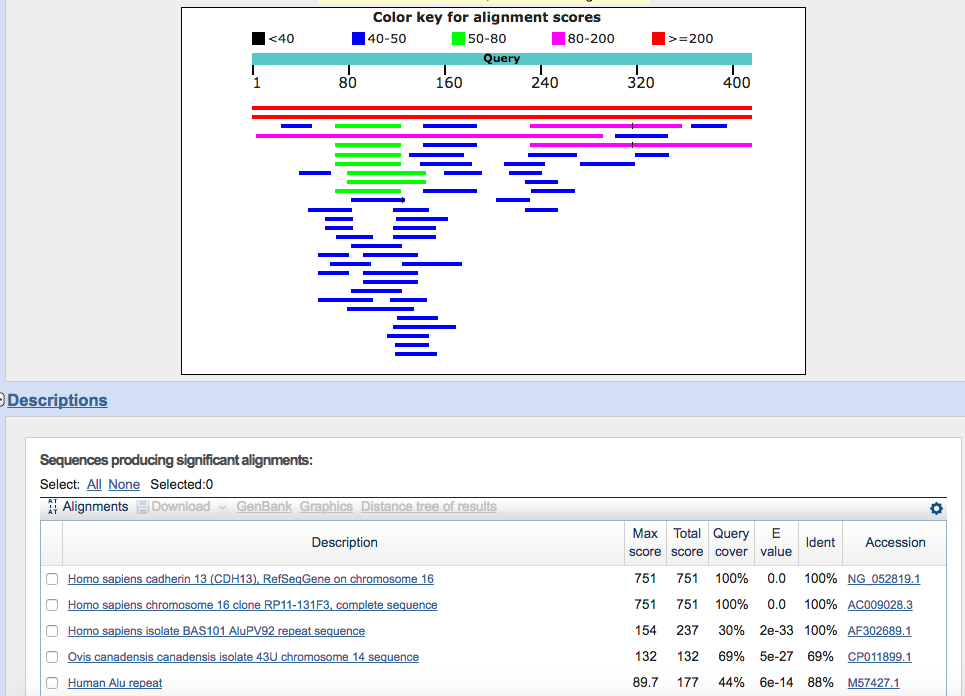
I primi due risultati li conosciamo già, si tratta delle regioni sul genoma umano di riferimento su cui si allineano i nostri primer.
Alcuni risultati però sono nuovi, in particolare soffermiamoci per primo sul risultato “Human Alu repeat”. Cliccando sul nome in blu potremo visualizzare l’allineamento:
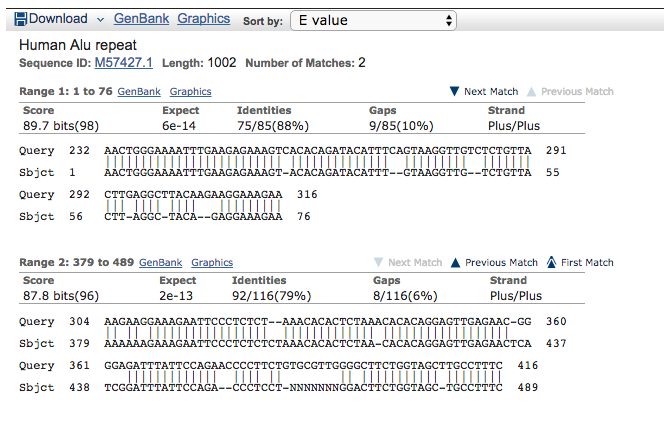
Come potete notare, e come era evidente già dal riassunto grafico, la nostra sequenza ottenuta dal genoma umano di riferimento si allinea sul tratto di sequenza Alu da 1 a 76, poi c’è un gap, e poi si allinea da 361 a 489.
Cosa ci sarà lì in mezzo?
Per scoprirlo andiamo a cercare informazioni più approfondite riguardo a questa sequenza cliccando sul suo codice identificativo “Sequence ID: M57427.1“. Si aprirà una finestra contenente tutte le informazioni in modo ordinato e ci concentreremo sulla sezione ” FEATURES “, di cui riporto uno screenshot
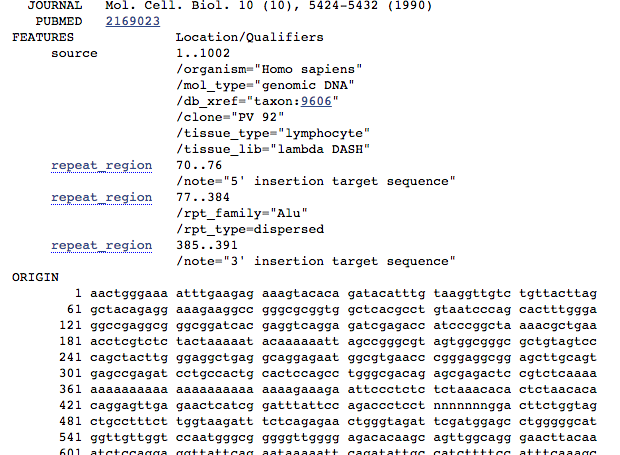
Features di interesse 1:
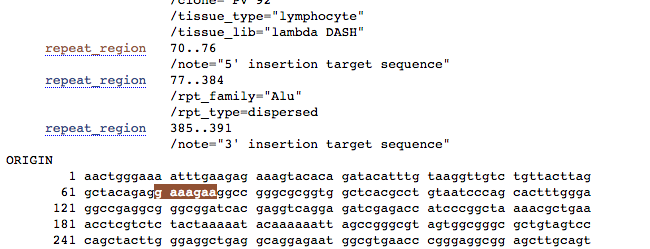
Features di interesse 2:
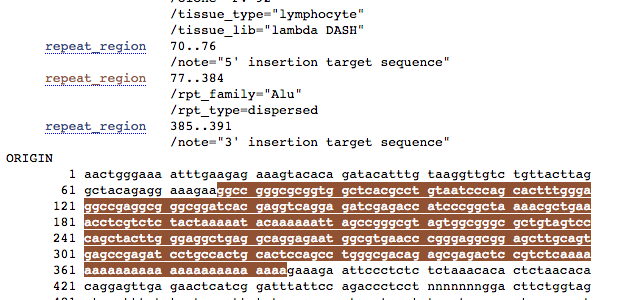
noti qualcosa nella regione 3′ della sequenza Alu evidenziata qui sopra?
vediamo insieme a cosa serve questa coda speciale cliccando su questo link:
https://www.dnalc.org/resources/animations/alu.html
Features di interesse 3:
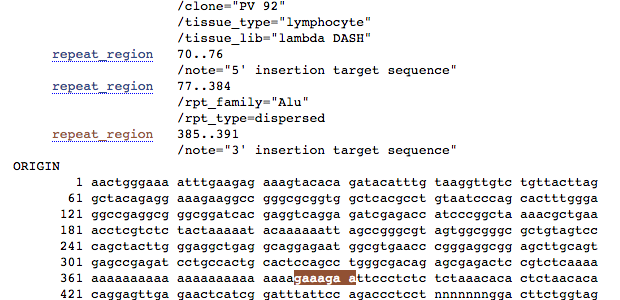
Abbiamo trovato la sequenza Alu e le sue sequenze fiancheggianti. Alla luce di questo, quello che avevi trovato sul genoma era l’allele contenente regioni fiancheggianti e sequenza Alu o la sola regione contenente le sequenze fiancheggianti?
Utilizzando i dati che hai a disposizione calcola la dimensione attesa dell’allele – e dell’allele +
Andiamo ora ad esplorare un altro risultato di allineamento interessante:
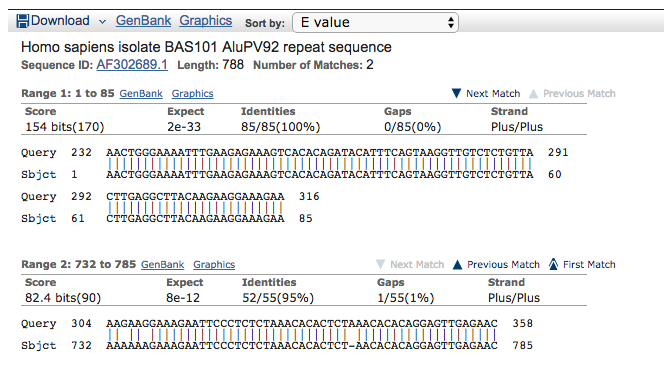
Clicchiamo anche in questo caso sul codice identificativo della sequenza per accedere alle informazioni più approfondite “Sequence ID: AF302689.1”
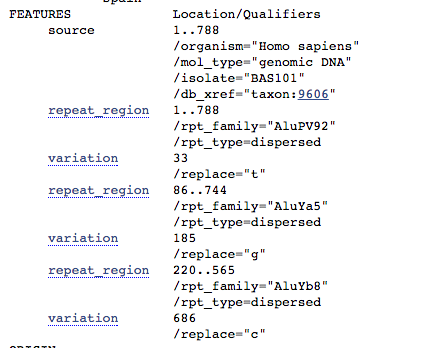
Osservate con attenzione e ditemi se notate qualcosa di interessante..
Le sequenze Alu nella popolazione umana
L’ultima parte della nostra analisi riguarderà la frequenza allelica e genotipica delle due varianti della regione Alu (Alu+ e Alu-) nelle popolazioni mondiali.
Per fare queste analisi comparative utilizzeremo il server www.BioServers.org del Dolan DNA Learning Center. Cliccate sul link e procediamo!
Il primo server che sceglieremo sarà l’ “ALLELE SERVER” ed entreremo senza registrarci, scegliendo semplicemente “ENTER”
Frequenze osservate e frequenze attese
Una volta dentro cliccheremo su “MANAGE GROUPS” per aprire una finestra pop-up con la lista degli esperimenti registrati, fatti da studenti e anche da gruppi di ricerca. Scegliendo nel menù a tendina in alto a destra “Classes” i dati in elenco saranno solo quelli prodotti da studenti durante le esercitazioni con il kit “Alu insertion polymorphism”.
Possiamo selezionare uno o più dei gruppi in elenco e poi cliccare su ok.
Nella pagina principale, selezionando uno dei gruppi con il quadratino a sinistra e cliccando su “OPEN” si aprirà una finestra di dialogo con tutti i dettagli disponibili sui campioni appartenenti a quel gruppo.
Sempre nella pagina principale, selezionando uno o più gruppi con il tondino a destra e poi cliccando “ANALYZE” possiamo procedere con l’analisi delle frequenze genotipiche. In quest’analisi vedrete la visualizzazione grafica dei dati raccolti e dei dati attesi. Il valore p-value rappresenta di nuovo la probabilità che i campioni siano diversi per caso. Più bassa è questa probabilità più alta è la confidenza con la quale potete dire che quei dati in effetti deviano dall’atteso a causa di qualche fenomeno di tipo biologico. Gli scienziati per convenzione usano come sogli di p-vaule 0,05, ovvero 95% di probabilità che il dato sia significativo.
Frequenze osservate in diverse popolazioni
In questa seconda parte dell’analisi compareremo le frequenze genotipiche e alleliche di diverse popolazioni.
Iniziamo di nuovo con la scelta dei dati da comparare cliccano su “MANAGE GROUPS” e aprendo la finestra pop-up con la lista degli esperimenti registrati. Questa volta però sceglieremo gli esperimenti sistematici e su grandi numeri fatti dai ricercatori professionisti scegliendo nel menù Select group type: Reference.
Come nel caso precedente selezioniamo alcune popolazioni cliccando sui quadratini a sinistra e confermando la scelta con “OK”
Nella pagina principale possiamo selezionare di nuovo i dati da analizzare, osservare le informazioni cliccando su “OPEN” e questa volta poi cliccheremo su “COMPARE” per vedere se ci sono differenze nelle frequenze alleliche fra le varie popolazioni.
Referenze
Il materiale utilizzato per questa esercitazione è stato predisposto dal DOLAN DNA Learning Center. Questa esercitazione in aula virtuale può essere accompagnata da una sezione sperimentale utilizzando i protocolli preparati dal DOLAN DNA Learning Center e distribuiti da Carolina sotto forma di Kit completi acquistabili online oppure da Biorad, sempre come Kit completi acquistabili online.
Personalmente, avendoli testati entrambi, consiglierei per gli utenti in Italia il kit biorad. La ragione più semplice riguarda la praticità di ordinare un prodotto distribuito in Italia evitando così di dover seguire lo sdoganamento dei prodotti ed evitando i fastidiosi e purtroppo frequenti inconveniente con il corriere Fedex (le chances di ricevere il prodotto scongelato e deteriorato dal caldo sono molto alte). Inoltre nelle mie mani (o meglio in quelle degli studenti che con me hanno lavorato) il protocollo di estrazione del DNA fornito da Biorad è più robusto.





Pingback: La bioinformatica è uno strumento per l’insegnamento della biologia – Sperimentando